Project Manager:
Prof. Maria Fyta, PhD
NanoMLmatDesign: Computational design of complex materials: from nanopores to alloys
Principal Investigators:
Prof. Maria Fyta, PhD
Affiliation:
University of Stuttgart
HPC Platform used:
NHR@KIT: HoreKa
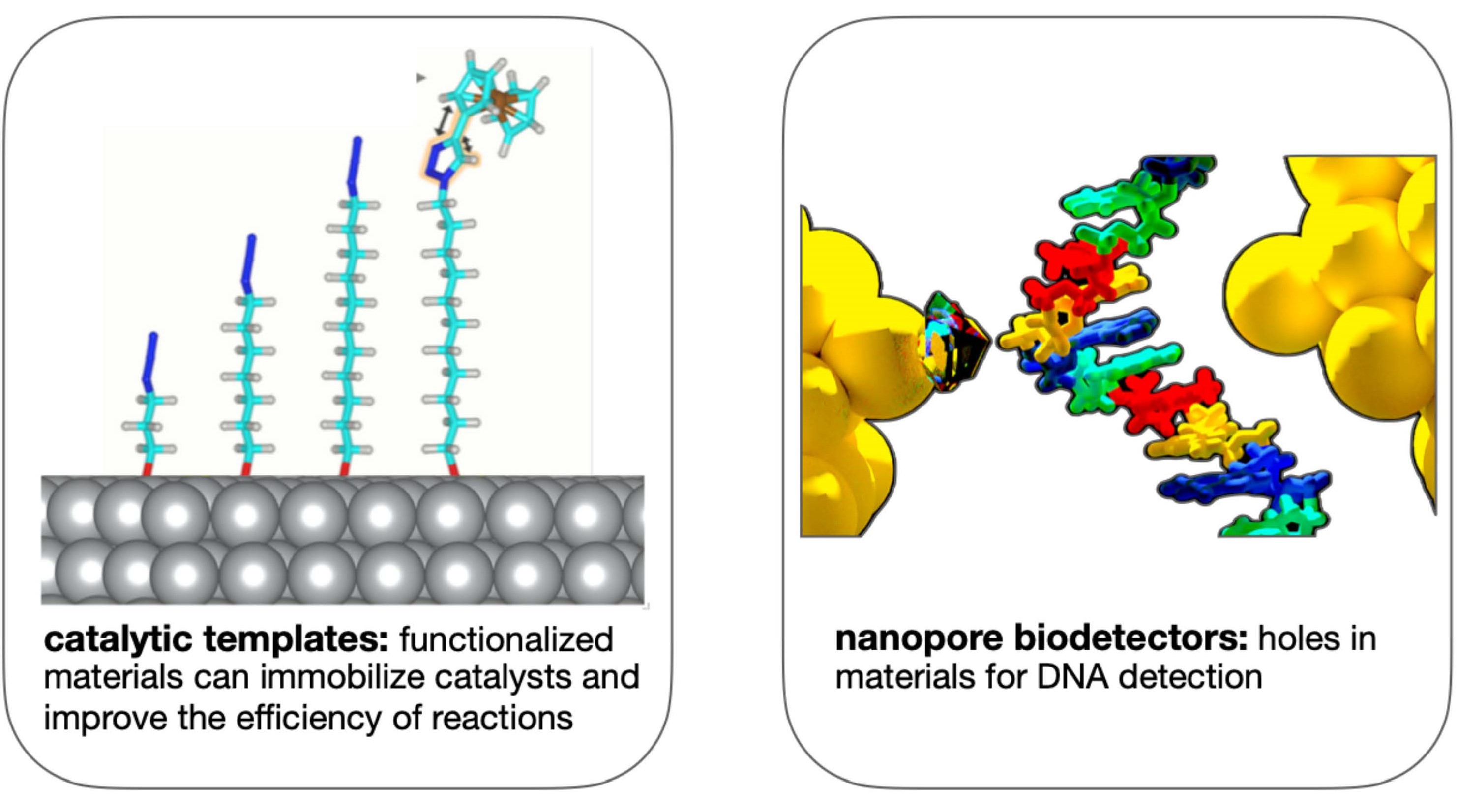
An optimum and selective materials design based on computational approaches is essential in order to avoid time-consuming and expensive experiments and drive novel applications in sensing, catalysis, and electronic components. Within this concept, (a) the optimization of nanoporous materials made of functionalized gold surfaces with self-assembled monolayers, as well as (b) the design of alloying materials were investigated. The investigations are directed towards (a) heterocatalysis and biosensing, as well as (b) alloys for strong and highly conducting electronic components were seeked. This research was performed using quantum-mechanical and atomistic simulations occasionally in combination with Machine Learning approaches.