Data-Driven Virus Discovery: Characterizing Viromes and Increasing Pandemic Preparedness
- Principal Investigators:
- Prof. Dr. Chris Lauber, Dr. Stefan Seitz
- Project Manager:
- Prof. Dr. Chris Lauber
- additional Affiliation:
- Cluster of Excellence RESIST, Hannover Medical School Helmholtz Corona Virus Pathogenesis (CoViPa) research network European Virus Bioinformatics Center
- HPC Platform used:
- NHR@TUD Barnard, Romeo, Julia, Alpha
- Project ID:
- p_sra
- Date published:
- Researchers:
- Nicolai Böker
- Introduction:
- Data-Driven Virus Discovery (DDVD) is revolutionizing the way novel viruses are discovered. Being independent of the collection and processing of biological samples, DDVD allows for screening massive amounts of next generation sequencing (NGS) data for the presence of known and unknown viral genome sequences. We utilize DDVD to analyze 1+ million of public NGS datasets from the Sequence Read Archive (SRA) and find 150+ thousand sequences of viral origin. We use these data to assess the risk of spillover into humans across the RNA viruses and to study various aspects of viral evolution across geologic time scales.
- Body:
-
Introduction
Viruses are ubiquitous, affecting all life forms. They have been associated with various diseases and regularly cause epidemics or pandemics in animals and humans. Despite their broad relevance, it is generally accepted that a major portion of the natural diversity of viruses, in particular concerning numerous understudied host groups, is still unknown, a notion that is reinforced by a steady inflow of newly discovered viruses. Novel viruses are typically identified through their gene or genome sequences in times of continuously advancing sequencing technologies, drastically outpacing other forms of virus characterization. Data-Driven Virus Discovery (DDVD) approaches offer a way to mine these sequencing data for known and novel viruses.Methods
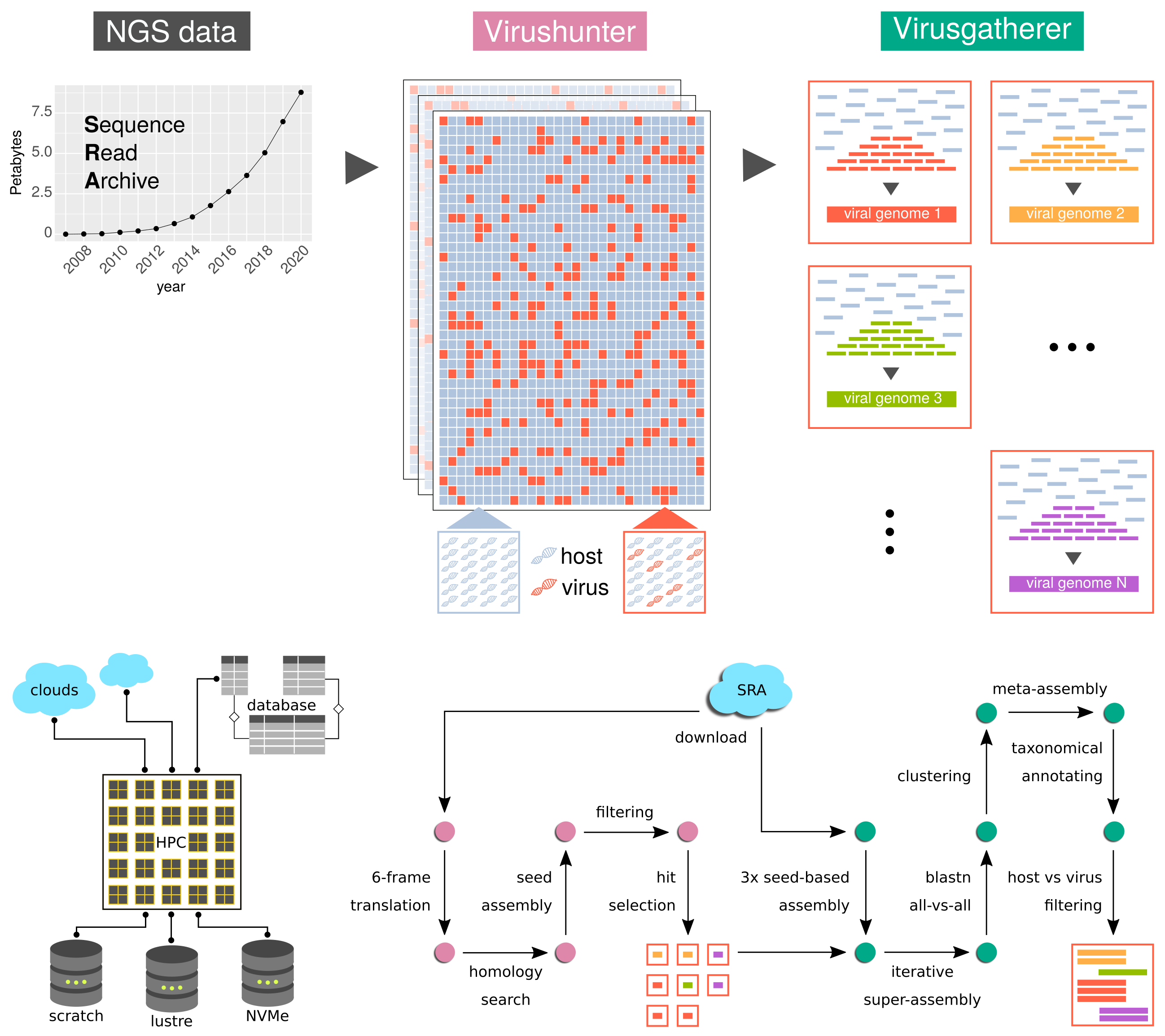
We have developed a DDVD approach involving a high-performance computing workflow for the discovery of viral sequences in unprocessed next generation sequencing (NGS) data from the Sequence Read Archive (SRA) repository. Our workflow is highly parallelized, involves the download and temporary storage of hundreds of Terabytes of SRA data and proceeds in three major steps (Figure 1). First, we efficiently screen the raw sequencing reads for significant similarity to viral marker genes (Virushunter stage). This involves a sensitive sequence homology search based on profile Hidden Markov Models (pHMMs). We subsequently conduct a targeted viral genome assembly (Virusgatherer stage) for those SRA data sets identified in the first stage. The identified viruses are then assigned to taxonomic groups in the last stage.Results
We have so far screened more than a million SRA data sets covering the full spectrum of available eukaryotic transcriptomes and additional genomes, including a subset of human SRA experiments. We have discovered more than 150 thousand of known and unknown RNA and DNA virus sequences, building a catalogue of viral genomic diversity that forms the basis for subsequent studies.
Among our discoveries are (i) the non-enveloped nackednaviruses, which are the closest relatives of the enveloped hepatitis B viruses (HBVs) and that allowed us to study the emergence of the HBV envelope gene several hundred million years ago, (ii) ambiviruses which are hybrids of RNA viruses and viroids, (iii) anelloviruses with putative associations with human diseases, (iv) bisegmented coronaviruses that encode the replicative and structural proteins on two separate genome segments and which offer new insights into the exchange of the Spike gene between different coronaviruses and coronaviruses and related tobaniviruses, (v) giant nidoviruses that have the largest known RNA genomes up to 64 kilobases, enabling us to study constraints of RNA genome evolution, and (vi) various novel proteins domains of currently unknown enzymatic or other function encoded by the discovered viral genomes, which will improve our understanding of virus-host interactions at different stages of the viral life cycle.Outlook
We will continue our screen of human SRA experiments and will regularly update our analysis of other eukaryotes. We will aim at correlating differences in virome composition between individuals of a host species with phenotypes, including human diseases, where available. We furthermore seek to develop a risk score for zoonosis to assess the likelihood of viral spill-over from animals into humans. We hypothesize that such a score will be instrumental for predicting future pandemic viruses. Moreover, we will study novel protein domains encoded by the discovered viral genomes by comparative genomics and 3D structural modelling.References
1. Neuman BW, Smart A, Gilmer, O, Smyth R, Vaas J, Böker N, Samborskiy DV, Bartenschlager R, Seitz S, Gorbalenya AE, Caliskan N, Lauber C. Giant RNA genomes: roles of host, translation elongation, genome architecture and proteome in nidoviruses. PNAS. 2025. In press
2. Lauber C, Zhang X, Vaas J, Klingler F, Mutz P, Dubin A, Pietschmann T, Roth O, Neuman BW, Gorbalenya AE, Bartenschlager R, Seitz S. Deep mining of the Sequence Read Archive reveals major genetic innovations in coronaviruses and other nidoviruses of aquatic vertebrates. PLoS Pathog. 2024 Apr 22;20(4):e1012163. doi: 10.1371/journal.ppat.1012163. PMID: 38648214; PMCID: PMC11065284.
3. Chong LC, Lauber C. Viroid-like RNA-dependent RNA polymerase-encoding ambiviruses are abundant in complex fungi. Front Microbiol. 2023 May 12;14:1144003. doi: 10.3389/fmicb.2023.1144003. PMID: 37275138; PMCID: PMC10237039.
4. Lauber C, Seitz S. Opportunities and Challenges of Data-Driven Virus Discovery. Biomolecules. 2022 Aug 4;12(8):1073. doi: 10.3390/biom12081073. PMID: 36008967; PMCID: PMC9406072.
5. Lauber C, Seifert M, Bartenschlager R, Seitz S. Discovery of highly divergent lineages of plant-associated astro-like viruses sheds light on the emergence of potyviruses. Virus Res. 2019 Jan 15;260:38-48. doi: 10.1016/j.virusres.2018.11.009. Epub 2018 Nov 16. PMID: 30452944.
6. Lauber C#, Seitz S#, Mattei S, Suh A, Beck J, Herstein J, Börold J, Salzburger W, Kaderali L, Briggs JAG, Bartenschlager R. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe. 2017 Sep 13;22(3):387-399.e6. doi: 10.1016/j.chom.2017.07.019. Epub 2017 Aug 31. PMID: 28867387; PMCID: PMC5604429.
# equal contribution
- Institute / Institutes:
- TWINCORE Centre for Experimental and Clinical Infection Research German Cancer Research Center (DKFZ)
- Affiliation:
- Hannover Medical School + Heidelberg University
- Image:
-